はじめに
医療機器のISO13485審査(QMS調査)において、数多くの企業の審査に携わってきた経験から、指摘が多い項目Top 10とその対策についてお話しします。19年間の主任審査員としての経験を通じて、特に指摘が多かった項目に焦点を当て、実践的な改善のポイントをご紹介します。
指摘が多かった項目及び指摘内容は、あくまでも筆者の審査経験からということを、ことわっておきます。
第1回目は、Top1~3でしたが、第2回は、Top 4~6について説明します。Top4からTop6は次の3項目ですが、これらは指摘数では、ほぼ横並びです。
- Top 4; ISO 13485/7.5.6 製造及びサービス提供に関するプロセスの妥当性確認
- (QMS 省令 第45条 製造工程等のバリデーション)
- Top 5; ISO 13485/8.2.6 製品の監視及び測定 (QMS 省令 第58条 製品の監視及び測定)
- Top 6; ISO 13485/8.4 データの分析 (QMS 省令 第61条 データの分析)
No.4:ISO 13485/7.5.6 製造及びサービス提供に関するプロセスの妥当性確認(QMS 省令 第45条 製造工程等のバリデーション)に対する指摘とその対策
この妥当性確認(バリデーション)に対する要求は、後の検査でその確かさを検証(検査)できない工程が該当する場合に適用されます。
例えば、溶接作業の確かさを後で確認するには、破壊強度を測定するしかなく、破壊するとその製品は使用できなくなってしまいます。鉄橋建設において、溶接部分があったとして、その強度の保証の根拠は、この溶接工程(プロセス)の妥当性確認(バリデーション)の結果ということになります。
ただし、その鉄橋の強度の保証は、鉄橋建設時の作業が、妥当性確認(バリデーション)の結果で、定められた条件どおりに作業されるということが前提となります。
指摘事項の具体例
(1) バリデーションの結果が作業手順書(製造作業条件)に反映されていない
バリデーションの目的は、バリデーションの結果を、そのプロセスの作業条件に反映させることです。
しかしながら、作業手順書(溶接標準条件)が、バリデーション(溶接強度試験など)の結果で得られた作業条件になっていませんでした。
(2) バリデーションが必要となる工程で、バリデーション結果の未記録又は未実施
バリデーションの記録が維持されていない、またはバリデーションを実施していませんでした。
(3) 規格で要求される項目の未文書化
ISO 13485/【7.5.6 製造及びサービス提供に関するプロセスの妥当性確認】には、下記の要求があります。
組織は,次を含むプロセスのバリデーションの手順を文書化する。
a) プロセスのレビュー及び承認のために定めた判断基準
b) 設備の認定及び要員の資格認定
c) 特定の方法,手順及び判断基準の使用
d) 適切な場合,サンプルサイズの根拠となる統計的手法
e) 記録に関する要求事項
f) 再バリデーションの判断基準を含む,再バリデーション
g) プロセスに対する変更の承認
ISO 13485の審査において、a)~g)のいずれかの手順が文書化されていなかった場合の、不適合です。
(4) 製造に使用したコンピュータソフトウェアのバリデーション未実施
例えばNCプログラム、(7.5.8で要求されている)機器固有識別(UDI)のラベル印刷用ソフトウェア又は3次元プリンタを用いて製品を作成する場合のソフトウェアは、バリデーションを実施する必要がありますが、そのバリデーションを実施していなかった場合の不適合です。
逸脱(不適合)原因とその対策
(1) バリデーションの結果が作業手順書(製造作業条件)へ反映されていない
原因は、バリデーションの目的の理解不足のようでした。ISO 13485の認証を得る目的として、バリデーションを実施して、その記録は維持されていました。しかしバリデーションの本来の目的を理解していなかったために、作業手順書へ落とし込みことを怠っていたため、生じた不適合でした。
対策としては、バリデーション計画書などに、要求事項を関係作業手順書などへ反映させることを明記しておくことなどでしょう。または、作業手順書の作業条件に根拠となったバリデーション報告書番号及び報告書名などを記載するようにしておくことなどが考えられます。
(2) バリデーションが必要となる工程で、バリデーション結果の未記録又は未実施
原因としては、ISO 13485の認証を取得する以前から、定められた条件で作業を継続していることがあげられます。別の原因として、その作業設備メーカーの推奨条件をそのまま採用していることなどがあげられます。
前者は、改めて現在実施されている条件を中心として、条件を変化させてみてバリデーションを実施することでしょう。
後者についてですが、設備メーカーはあくまでも一般的な場合(溶接の場合、素材・厚み)についての条件を推奨している場合は多いので、自社の製品(溶接の場合、素材や厚みなど)の条件でバリデーションを実施することが必要です。
(3) 規格で要求される項目の未文書化
原因としては、規格の要求事項の理解不足などがあげられます。バリデーション計画及び実施結果に中に手順があれば。文書化された手順とみなせる場合があります。
規格の要求事項のd)には、「適切な場合,サンプルサイズの根拠となる統計的手法」の“適切な場合”ということわり書きがあります。この規格の“適切な場合”の説明が、「0.2 概念の明確化」に次のように記載されています。
要求事項が“適切な場合”という用語で特定された場合,組織が他の方法によることの正当性を示すことができなければ,その要求事項の適用は“適切”であるとみなされる。
組織が他の方法によることの正当性を示すことができれば、サンプルサイズの根拠に、統計的手法を使用しなくても良い事になります。
(4)製造に使用したコンピュータソフトウェアのバリデーション未実施
この要求事項は、ISO 13485/ 7.5.6のそれ以前に記載された要求事項とは、独立しているとみなされ、製造に使用したコンピュータソフトウェアであれば、すべてバリデーションが必要という要求です。
検査用コンピュータソフトウェアは、「7.6 監視機器及び測定機器の管理」の後半に記載され、ここも前段の記載とは独立した要求事項とみなされます。
製造用でも検査用でもないコンピュータソフトウェア、例えば購買用コンピュータソフトウェアのバリデーションは、4.1.6で要求されています。
重要なポイント
製造に関するプロセスの不適切なバリデーションは、医療機器のリスクに直結する場合もあると考えられます。また、バリデーションの結果で得られた作業条件は、必ずそのとおりに作業を実施しないと重要な結果を招きます。
最後に、医療機器ではありませんが、9名の死者を出した2012年12月2日発生の笹子トンネル天井板落下事故の事例について紹介しておきます。
----------------------------
見出し「そんなに簡単に抜けるとは」「ボルト調査・設計上は4トン以上」
笹子トンネルのボルトは、鉄製で深さ13センチの穴に埋め込まれている。天井をつり下げるため、1本あたり1.2トン程度の重さがかかるが、設計上はその4倍以上の重みに耐えることになっていた。(筆者注記;バリデーションを実施した結果と推察される。)
一方写真入りで、次の記載があった。
国交省の調査時に抜けたボルト。(写真掲載)
接着剤は先端から半分までしか届いていなかったとみられる。
(筆者注記;バリデーション実施結果で作成された作業条件どおりに作業を実施していなかったと考えられる。)
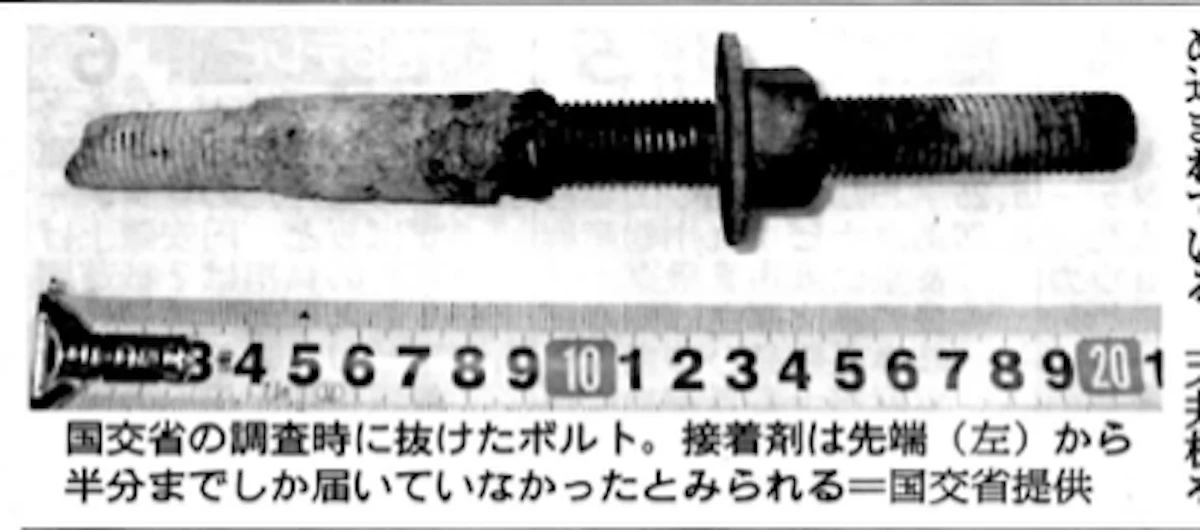
(2013年2月2日付け朝日新聞から引用)
No.5:ISO 13485/8.2.6 製品の監視及び測定 (QMS 省令 第58条 製品の監視及び測定)に対する指摘とその対策
この要求事項の項では、測定に使用した測定器の識別に関しての指摘(不適合)が、一番多かった。
指摘事項の例
(1) 使用した測定器番号などの未記録
ISO 13485/ 7.6の要求事項には、「適切な場合,記録には,測定活動の実施のために使用した試験機器を識別する。」という要求があります。測定器が2台以上あってどちらも任意に使用できる状態にある場合に、どの測定器を使用したかの識別の記録がなかった場合の指摘です。
(2) 重要検査項目の抜け
医療機器の品目仕様などに、仕様項目値が記載され、それがその医療機器にとって重要で必要な仕様であり、かつその仕様値範囲内に入っていないとリスクがある場合にも関わらず、その項目が検査されていなかったケースです。
要求事項「組織は、製品要求事項が満たされていることを検証するために、製品の特性を監視し、測定すること。」対する逸脱です。
(3) 抜取り検査におけるロットの定義が不明確
検査手順に、「製造ロットを確認し、抜取数規定表に従って検査台数を決定する。」と記載されていましたが、製造ロットの定義が具体的に規定されていないため、抜取り検査台数が、正しいかどうか判定できなかったケースです。検査者の判断によって、ロットの範囲を広くも狭くもできる状態だったケースです。
要求事項「監視及び測定は.計画し文書化された取り決め及び文書化された手順に従って,製品実現プロセスの適切な段階で実施 する。」という要求事項対して、その文書化された手順のおけるロットの定義がなく、実施段階でロットの特定が不適切と判断されたケースです。
逸脱(不適合)原因とその対策
(1)使用した測定器番号などの未記録
逸脱の発生原因は、規格の要求の意味・目的・狙いを深く理解していなかったこと、識別しないことのリスクを認識していなかったこと、品質マニュアルには、ISO 13485:2016の要求事項がそのまま記載されていたが、具体的に識別方法が確立されていなかったこと、ISO 13485:2016で新しく追加された要求事項であることなどが、挙げられます。
なぜ測定に使用した測定器の識別が必要であるか、それはISO 13485/7.6の要求「さらに,測定機器が要求事項に適合していないことが判明した場合には,組織は,その測定機器でそれまでに測定した結果の妥当性を評価し,記録する。組織は,その装置及び影響を受けた製品に関して,適切な処置をとる。」に関係しています。検査に仕様された測定器を識別していない場合、測定機器が要求事項に適合していないことが判明した場合、その適合していない測定器で測定した医療機器製品を特定できないことになります。
要求事項には、“適切な場合”とあります。これは測定器が1台しかなかった場合に識別は不要であることを意味しています。
(2) 重要検査項目の抜け
原因としては、被審査部門からの是正処置報告書には、設計・開発部門と製造部門への伝達方法が良くなかったと報告されていました。
一般的に設計・開発部門は、製品要求事項(製品開発インプット)から、その製品にとって何が重要か考えて、設計開発を実施します。しかし設計・開発が完了し、製造へ渡すときにそのような情報が欠如したケースです。
ISO 13485/7.3.4 設計・開発からのアウトプットに、「b) 購買,製造及びサービス提供に対して適切な情報を提供する。」というのがありますが、この適切な情報とは何かを今一度見直していくことが必要と考えられます。
(3) 抜取り検査におけるロットの定義が不明確
ロットとは、本来「同じ条件で製造される製品の製造数量、出荷数量の最小単位」という意味です。
それを複数のロットをまとめて、ロットと判断してしまうと、適切な抜取り検査を維持できなくなってしまいます。この不適合の原因は、ロットという意味とロットに対する抜取り検査の本来の目的・意味を明確にしていなかったために生じた不適合です。
対策としては、「同じ条件で製造される製品の製造数量単位」を明確にして、ロット番号を厳密に付けていくことなどが考えられます。
重要なポイント
このISO 13485/8.2.6 製品の監視及び測定は、医療機器製品が、使用者へ公表している仕様を満足しているかを保証する上で、重要な工程です。組織としては、十分に注意していることが、推定されますが、(1)の例のように、要求の中でも(副次的な)一部を見逃してしまうことが生じていました。ISO 13485の細部の要求に対しても適合するように、折に触れて例えば内部監査などにより確認していくことが重要と考えられます。
No.6:ISO 13485/8.4 データの分析 (QMS 省令 第61条 データの分析)に対する指摘とその対策
指摘事項の例
(1) データ分析記録に、規格で要求されている項目抜け
ISO 13485/8.4 データの分析で、要求している項目は下記である。
- a) フィードバック
- b) 製品要求事項への適合性
- c) 改善の機会を得ることを含む,プロセス及び製品の特性及び傾向
- d) 供給者
- e) 監査
- f) 適切な場合,附帯サービスの報告書
実際の審査で、不足していた項目は、「d)供給者」、「e)監査」、又は「f)附帯サービスの報告書」でした。
(2) 少ないデータによる分析
例えば、「d)の供給者」、「e)の監査」について、単年度(その年のみ)のシンプルなデータをマネジメントレビューで報告していたことをデータ分析と称していました。
ISO 13485の要求事項の目的・狙い「データ分析により改善へ結びつける」から言えば、例えば、5年間分などの多くのデータを用いて分類とか傾向をみて、改善へ結びつけることのできるデータ分析が求められます。
逸脱(不適合)原因とその対策
(1) データ分析記録に、規格で要求されている項目が抜け
e) 監査及びf) 適切な場合,附帯サービスの報告書が、2016年版で追加されていましたが、その対応が不十分だったという原因がほとんどでした。品質マニュアルには、新規要求を追加したが、データ分析の様式は、そのままにしておいたのが原因という組織もありました。規格改定の時には、改訂後の規格を単に、品質マニュアルに追加するだけでなく、その下位文書及び使っていた記録様式にまで影響がないか注意を要します。
(2) 少ないデータによる分析
「d)の供給者」、又は「e)の監査」について、単年度(その年のみ)の数少ない・シンプルなデータをマネジメントレビューで報告していたことをデータ分析と称した場合、なぜに不適合となるか、その根拠はどこにあるか?
ISO 13485/8.4の冒頭には、下記要求があります。
組織は,品質マネジメントシステムの適切性,妥当性及び有効性を実証するため,適切なデータを明確にし,それらのデータを収集し,分析するための手順を文書化する。
もし,このデータ分析によって,品質マネジメントシステムの適切性,妥当性又は有効性がないことが示された場合は,8.5で要求しているように,組織はこの分析を改善のためにインプットとして用いる。
品質マネジメントシステムの適切性,妥当性及び有効性を実証するためのデータは、単年度で十分か、単年度の例えば内部監査の当年度だけでは、データ分析ではなく、単なる分析と言えるでしょう。
当年度だけでも、かなり多くの不適合があって、それを部門別とか、ISO 13485のどの項目で不適合が多かったなどの分析を行っていれば、データ分析と言えるかも知れません。しかしながら例えば少数(例 5件以下)の不適合に対して、内部監査報告書をもってデータ分析と言えるのか? 少なくとも品質マネジメントシステムの適切性,妥当性及び有効性を実証するには、データ不足と言えるのではないでしょうか?
重要なポイント
データ分析の目的・意図・狙いは、ISO 13485/8.4の要求事項の中に記載されています。
データ分析の目的・意図・狙いは、
・「品質マネジメントシステムの適切性,妥当性及び有効性を実証するため」であり、
・このデータ分析によって,品質マネジメントシステムの適切性,妥当性又は有効性がないことが示された場合は、「8.5で要求しているように,組織はこの分析を改善のためにインプットとして用いる。」ことが重要です。
また月別、ロット別推移の製品合格率の分析を行っていて、急に不自然に合格率が極端に良くなった場合にも注意すべきです。原因調査の結果、その理由が明確になっていれば、問題のないのですが、全く不自然に極端に合格率があがった場合、世の中で良く問題になっているように、「データ改ざん」の可能性さえあります。特に製品を委託製造している場合に注意を要します。
QMS(品質マネジメントシステム)が、PDCAサイクルと言われますが、データ分析はこのPDCAのA(Action)が必要かどうかのC(Check)に該当します。
'/%3e%3c!--%20N%20monogram%20--%3e%3cpath%20d='M565%20206L485%20207L484%20512L244%20268L187%20326V665H267L268%20411L518%20665H565V206Z'%20fill='url(%23yn-navy)'/%3e%3c!--%20Accent%20square%20--%3e%3crect%20x='463'%20y='40'%20width='105'%20height='109'%20fill='url(%23yn-blue)'/%3e%3c/svg%3e)
.webp?w=1344&h=672&fm=webp&q=75)
%20(1).webp?w=800&h=450&fm=webp&q=75)




