はじめに
医療機器は人々の健康と生命に直接関わる製品であり、その製造と販売には厳格な規制が設けられています。日本では、「医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律(以下、「医薬品医療機器等法」)」によって、医療機器の製造・販売に関する規制が定められています。本記事では、医療機器の製造販売業と製造業の仕組みについて、わかりやすく解説していきます。
医療機器の定義・分類と製造販売を行うために必要なこと
医療機器とは
医薬品医療機器等法第2条第4項では、医療機器を以下のように定義しています:
「人若しくは動物の疾病の診断、治療若しくは予防に使用されること、又は人若しくは動物の身体の構造若しくは機能に影響を及ぼすことが目的とされている機械器具等(再生医療等製品を除く)であって、政令で定めるもの」
具体的には、以下のようなものが医療機器に該当します:
- 診断用機器(X線装置、超音波診断装置など)
- 治療用機器(手術器具、人工臓器など)
- 検査用機器(血圧計、体温計など)
- 予防用機器(滅菌器など)
また、医療機器の定義にある「機械器具等」の定義は、「機械器具、歯科材料、医療用品、衛生用品並びにプログラム、およびこれを記録した記録媒体」となっており、機械器具に組み込まれているプログラム、ならびに機械器具を伴わない単体プログラムも医療機器として位置づけられています。
医療機器の分類とリスクレベル
医療機器は、人体に与えるリスクの程度によって以下の3つのクラスに分類されます:
- 一般医療機器(クラスⅠ)
- リスクが極めて低い医療機器
- 例:医療用メス、X線フィルム、歯科技工用品
- 管理医療機器(クラスⅡ)
- リスクが比較的低い医療機器
- 例:電子式血圧計、電子内視鏡、画像診断装置
- 高度管理医療機器(クラスⅢ・Ⅳ)
- リスクが比較的高い、または生命の危険に直結する可能性がある医療機器。このうち、生命の危険に直結する可能性がある医療機器はクラスⅣとされています。
- クラスⅢの例:透析装置、人工骨
- クラスⅣの例:ペースメーカー、人工心臓弁
医療機器に係る業態
医療機器を業として取り扱う場合は、その業に応じた規制がその企業や事務所に適用され、国や都道府県庁等の許可や登録が必要となります。
- 製造業登録:医療機器の設計、主たる製造行為、滅菌、最終保管
- 製造販売業許可:医療機器を市場に出荷
- 販売業・貸与業許可/届:病院、診療所等への医療機器の販売・貸与
- 修理業/許可:医療機器の修理
製造販売業者は、医療機器を市場に出荷することはできますが、病院、診療所等に販売、貸与を行うことはできません。
また、各業態の許可申請を行う前に、業者コードを取得する必要もあります。業者コードの発行は各都道府県庁に申請します。
医療機器の製造販売を行うために必要なこと
日本において、医療機器を製造販売していくためには、製造販売業の許可を得る以外に製造販売する医療機器の許認可取得・届出を行う必要があります。
製造販売承認/認証/届出:製品の有効性・安全性等の審査と製造販売体制の調査
製造販売する医療機器の有効性・安全性を審査するとともに、製造販売業者の責任体制の審査および製造業者の製造方法・管理体制の審査を行うQMS適合性調査を実施します。この医療機器の審査ならびにQMS適合性調査による適合確認ができた場合に、医療機器の製造販売承認/認証が得られます。
製造販売承認を受ける場合の流れ
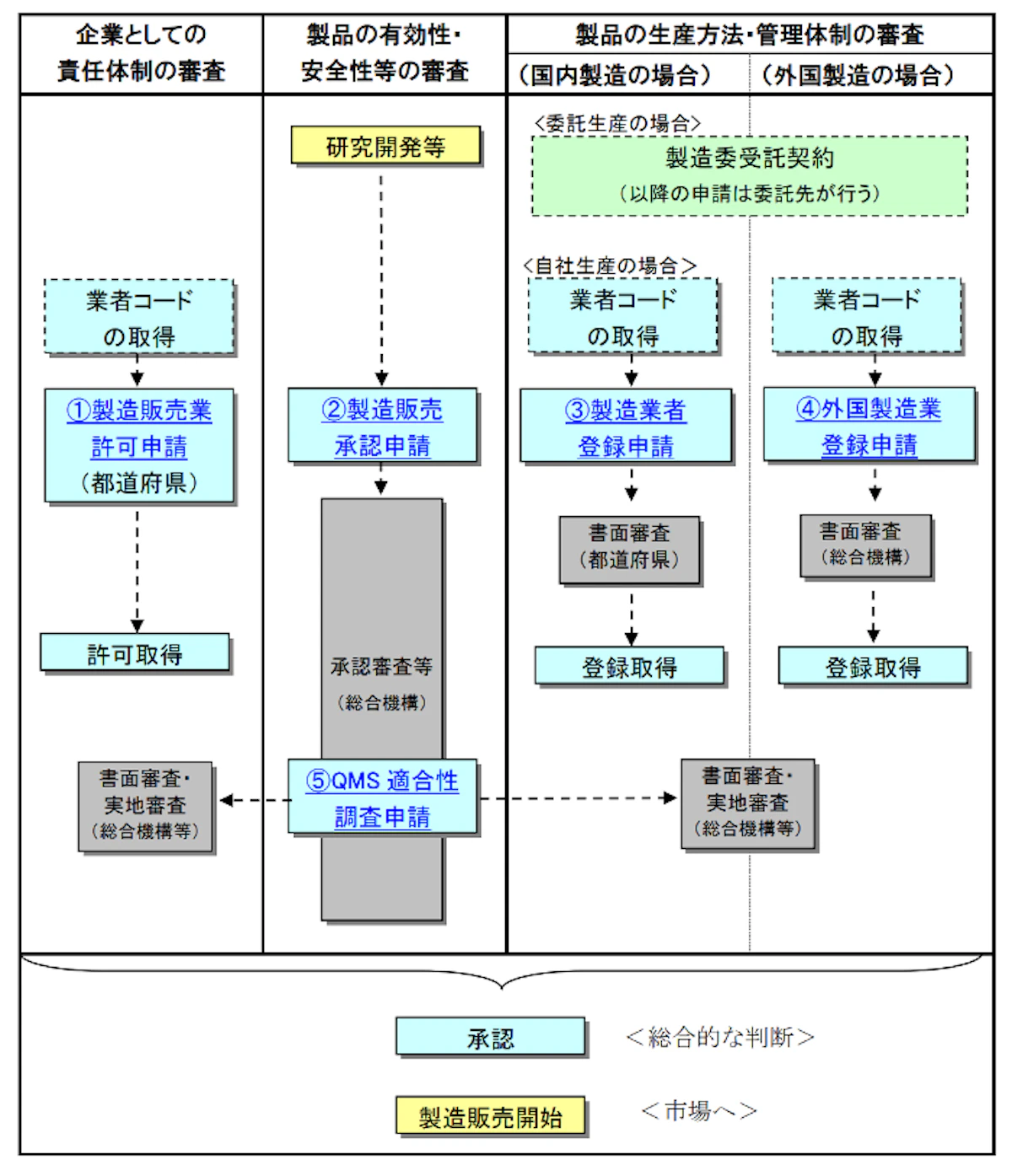
引用:PMDA医療機器の製造販売手順について
品目ごとの承認・認証・届出制度
製造販売を行う医療機器のクラス分類(リスクの大きさ)によって、PMDAによる承認が必要か、第三者機関からの認証が必要か、届出のみでよいのか、を判断することになります。
- 高度管理医療機器(クラスⅢ・Ⅳ)
- 原則としてPMDAへの承認申請が必要
- 臨床データの提出が必要な場合がある
- QMS適合性調査が必要
- 管理医療機器(クラスⅡ)
- 認証基準がある場合は第三者認証機関による認証
- 認証基準がない場合はPMDAへの承認申請
- QMS適合性調査が必要
- 一般医療機器(クラスⅠ)
- PMDAへの届出のみ
- 原則としてQMS適合性調査は不要
製造販売業について
製造販売業の定義と役割
製造販売業は日本独自の業態であり、医療機器を市場に供給する上で中心的な役割を果たします。
日本の場合、多くの医療機器が輸入されているため、実際の医療機器製造業者と日本における製造販売の組織が違うことが多々発生します。日本で製造販売される医療機器の有効性、安全性が日本の基準を満たしていること、日本で発生した品質や安全性に問題が発生した場合に、その責任の所在を明確にするとともに、患者の安全性を遅延なく確保することが製造販売業設置の目的の一つとなっています。
製造販売業者は、自社で製造するか外部委託するかにかかわらず、市場に出荷される医療機器の品質と安全性に対して最終的な責任を負います。
その責務は製品の出荷時点だけでなく、市販後の安全管理にまで及びます。
製造販売業は、平成17年度から新設された許可制度で、以下の特徴があります:
- 市場に出荷される医療機器の品質と安全性に対する最終的な責任を負う
- 製品の副作用情報やクレーム情報を収集・分析する
- 問題発生時の回収等の対応を行う
- 製造所の品質管理体制を管理監督する
許可の種類
製造販売業の許可は、取り扱う医療機器の最高リスクレベルによって3種類に分かれます:
- 第一種医療機器製造販売業
- 高度管理医療機器(クラスⅢ・Ⅳ)を取り扱う場合
- 下位クラスの医療機器も取り扱い可能
- 第二種医療機器製造販売業
- 管理医療機器(クラスⅡ)を取り扱う場合
- 一般医療機器も取り扱い可能
- 第三種医療機器製造販売業
必要な体制
製造販売業には、以下の体制整備が必要です:
- 総括製造販売責任者の設置
- 製造販売する医療機器の品質、安全性の全体的な管理監督を行う責任者の設置
- 品質管理体制
- QMS(Quality Management System)体制省令に適合した体制の整備
- QMS省令に適合した品質管理監督システムの構築
- 国内品質業務運営責任者の設置
- 安全管理体制
- GVP(Good Vigilance Practice)省令に適合した体制の整備
- GVP省令に適合した安全管理業務手順の構築
- 安全管理責任者の設置
製造販売業者に求められる3つの重要な責任者(三役)
この三名の責任者は、「製造販売三役」と呼ばれる重要な責任者となります。これは単なる役職ではなく、医療機器の品質と安全性を確保するための重要な管理体制の要となります。
1. 総括製造販売責任者
- 製造管理・品質管理・製造販売後安全管理の総責任者
- 資格要件は取り扱う医療機器のクラスによって異なります:
<第一種・第二種医療機器製造販売業の場合>
- 大学等での物理学、化学、生物学、工学などの専門課程修了者
- 旧制中学・高校などでの専門課程修了後、3年以上の実務経験者
- 5年以上の実務経験後、厚生労働大臣登録の講習修了者
<第三種医療機器製造販売業の場合>
- 要件が緩和され、旧制中学・高校などでの関連科目修了者
- 関連科目修得後3年以上の実務経験者
- 業務内容:
- 製品の出荷の決定その他の製造管理及び品質管理に係る業務を統括しこれに責任を負う
- 業務を円滑に行うために必要があると認めるときは、製造販売業者等、管理監督者その他の当該業務に関して責任を有する者に対し文書により必要な意見を述べ、その写しを5年間保管する
- 国内品質業務運営責任者を監督する
- 管理責任者及び国内品質業務運営責任者の意見を尊重する
- 製造管理または品質管理に関係する部門と製造完売後暗線管理基準に規定する安全管理統括部門との密接な連携を図らせる
2. 国内品質業務運営責任者
- 国内における品質管理業務の責任者
- 主な要件:
- 品質管理業務に3年以上の従事経験
- 国内の品質管理業務を適切に遂行できる能力
- 販売部門に属さないこと
- 業務内容:
- 国内の品質管理業務を統括する
- 国内の品質管理業務が適正かつ円滑に行われていることを確認する
- 国内に流通させる製品について、市場への出荷の決定をロット毎に行い、その結果及び出荷先当市場への出荷の記録を作成する
- 国内に流通させる製品について、当該製品の品質に影響を与える恐れのある製造方法、試験検査方法等の変更がなされる場合にあっては、当該変更が製品の品質に重大な影響を与える恐れがある場合には、速やかに管理責任者及び総括製造販売責任者に対して文書により報告し、必要かつ適切な措置が採られるようにする
- 国内に流通させる製品について、品質情報の収集・評価・措置(回収を含む)を実施する
- 製造所との連携による品質確保を行う
- 安全管理措置に関する情報を知った時は、安全管理統括部門に遅延画文書で提供する
3. 安全管理責任者
- 製造販売後の安全管理業務の責任者
- 要件は製造販売業の種類により異なります:
<第一種医療機器製造販売業の場合>
- 安全管理統括部門の責任者であること
- 安全確保業務に3年以上の従事経験
- 安全確保業務を適切に遂行できる能力
- 販売部門に属さないこと
<第二種・第三種の場合>
- 安全確保業務を適切に遂行できる能力
- 販売部門に属さないこと
- 業務内容
- 安全確保業務の統括
- 安全確保措置が適正かつ円滑に行われているかを確認し、その記録を作成、保存する
- 安全確保業務について必要であると認められた場合に、総括製造販売責任者に対し文書で意見を述べ、その写しを保存する
製造販売業の許可要件
製造販売業の許可を取得するためには、以下の基準を満たす必要があります:
- QMS体制省令への適合
- 製品の品質管理システムの確立
- 品質管理手順の文書化
- 組織体制の整備
- 製造所の管理監督体制の構築
- GVP省令への適合
- 安全管理情報の収集・評価
- 安全確保措置の実施
- 安全管理情報の伝達
- 安全管理教育訓練の実施
- 人的要件
- 前述の製造販売三役の設置
- 各責任者の資格要件の充足
- 必要な人員の確保
製造業について
製造業の定義と役割
製造業は、医療機器の実際の製造を担う業態ですが、医療機器の設計、滅菌、市場への出荷判定を行う製品保管も製造業と分類されています。製造販売業者の管理監督のもと、品質の確保された製品を製造する責任を負います。製造業の登録は製造所ごとに、それぞれの製造所で適切な製造管理と品質管理を行う体制を整える必要があり、以下の特徴があります:
- 医療機器の製造に特化した登録制度
- QMS省令に準拠した品質管理監督システムの構築
- 製造販売業者の管理監督下で品質管理を行う
- 製造業の登録のみでは市場への出荷はできない
登録の要件
製造業の登録には、以下の要件を満たす必要があります:
- 申請者の要件
- 欠格事由に該当しないこと
- 法人の場合は、その業務を行う役員も同様
- 責任技術者の設置
責任技術者の要件
責任技術者には、以下のいずれかの資格要件が必要です:
- 一般的な要件
- 大学で物理学、化学、生物学などの専門課程を修了
- 旧制中学・高校で専門課程修了後、3年以上の実務経験
- 5年以上の実務経験後、厚生労働大臣の登録を受けた講習修了
- 一般医療機器のみを製造する場合の要件
- 要件が若干緩和され、専門課程修了のみでも可能
- 関連科目修得後、3年以上の実務経験でも可
製造業の登録が必要な工程
医療機器の種類や特性に応じて、以下の工程で事業所ごとに製造業の登録が必要となります:
- 設計
- プログラム医療機器
- クラスⅠを除く医療機器
- ただし、製造販売業の主たる事務所と同一の場合は不要
- 主たる組立てその他の主たる製造工程
- 最終製品の組立て
- 重要部品の組立て
- 品質に重大な影響を与える工程
- 滅菌
- 滅菌が必要な医療機器の場合
- 滅菌バリデーションの実施が必要
- 国内における最終製品の保管
- 市場への出荷判定時に製品を保管している施設
- 品質管理上重要な保管条件の管理が必要
製造業者の責任体制
- 医療機器責任技術者の設置
- 製造所ごとに設置が必要
- 製造の実地場所において製造管理を担当
- 資格要件は製造する医療機器のクラスによって異なる
- 製造管理及び品質管理の責任者として機能
- 製造管理体制の整備
- 品質管理監督システムの導入
- 製造手順の文書化
- 製造記録の保管
- 製造設備の管理
- 製造環境の管理
- 従業員の教育訓練
- 品質管理体制の整備
- 品質管理基準の設定
- 試験検査の実施
- 品質記録の保管
- 不適合品の管理
まとめ
医療機器の製造販売業・製造業は、人々の健康と命を支える重要な責務を担っています。そのため、法規制による厳格な管理体制の確立が求められ、特に製造販売業者の三役体制は、製品の品質と安全性を確保するための核心的な仕組みとなっています。
これらの規制は、一見すると煩雑に見えるかもしれません。しかし、これらは全て医療機器の品質と安全性を確保し、最終的には患者さんの健康と命を守ることにつながっています。医療機器ビジネスに関わる企業には、こうした規制の本質的な意義を理解し、適切な体制を整備することが求められています。
'/%3e%3c!--%20N%20monogram%20--%3e%3cpath%20d='M565%20206L485%20207L484%20512L244%20268L187%20326V665H267L268%20411L518%20665H565V206Z'%20fill='url(%23yn-navy)'/%3e%3c!--%20Accent%20square%20--%3e%3crect%20x='463'%20y='40'%20width='105'%20height='109'%20fill='url(%23yn-blue)'/%3e%3c/svg%3e)